 Figure 1. Examples of enediyne natural products.
Figure 1. Examples of enediyne natural products.
The unprecedented structural elucidation of the enediyne-containing natural product antibiotics calicheamicin and esperamicin families in 1987, and later dynemicin A (Figure 1), demonstrated the biological importance of the reactive (Z)-1,5-diyne-3-ene functional unit. These natural products are isolated by microbial fermentation and exhibit astonishing activity against a variety of cancers, including P338 leukemia and B16 melanoma at doses of 0.15 mg/kg. The mechanisms of action of these agents are initially based on minor groove interaction and intercalative binding to DNA. Calicheamicin g1I and esperamicin A1 accomplish this by side chain (aryl tetrasccharide/substituted aromatic ring, fucosyl anthranilate, respectively) interactions with the minor goove, while the anthraquinone of dynemicin-A intercalates into the DNA double helix. The second step involves reductive activation of either the trisulfides of calicheamicin g1I and esperamicin A1, or the anthraquinone of dynemicin-A, which generates the toxic 1,4-phenyl diradical that performs the C-4' or C-5' H-atom abstraction from the deoxyribose ring, leading to single and double strand breaks. Unfortunately, the mild activation temperatures for enediyne cyclization (<37° C) lead to toxicity, and combined with the difficulties of synthetic accessibility, discouraged their further development. However, monoclonal antibody conjugates of calicheamicin have been in clinical trials for the past decade to treat AML and CD-33 positive Leukemia. While these seasoned agents begin to see applications, new enediyne structures continue to be discovered (tiancimycins, sealutomicins), suggesting that these constructs will continue to emerge and when combined with modern medical technology advances, will ultimately realize their antibiotic potential.
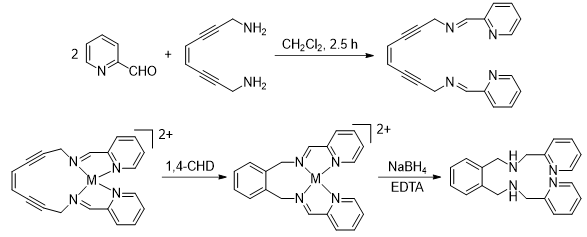 Scheme 1. Ambient temperature Bergman cyclization
Scheme 1. Ambient temperature Bergman cyclization
via reactive metalloenediyne constructs.
Minimized Metalloenediyne Structures
Retreating to conceptual development of activation mechanism alternatives, recent advances in the use of metal ions to control thermal Bergman cyclization via geometric structural changes has led to revitalized potential for small, easily prepared enediyne constructs. It is important to note, metal-binding to these enediynes modulates their cyclization temperature by 200°C relative to uncomplexed enediyne, far exceeding thermal control by simple carbon or organic functionalities.
Because enediynes undergo thermally-controlled reactions, hyperthermal activation may be a key towards realization of their full clinical potential. Hyperthermia is used as a cancer modality in some clinics, particularly as an adjuvant to radiotherapy and chemotherapy. Hyperthermia results in time-temperature dependent cell lethality, but even sub-lethal heating may enhance the effects of chemotherapeutic agents, or sensitize cells to ionizing radiation. While the mechanism of potentiation remains to be elucidated, heat induces many structural/functional alterations in cells, particularly in proteins which may become denatured. Our initial demonstration that metal-bound enediyne constructs are viable for hyperthermal activation came by way of several design iterations which revealed that the bis(pyridyl)-diiminoenediyne ligand, (Z)-N, N'-bis[1-pyridin-2-yl-meth-(E)-ylidene]oct-4-ene-2,6-diyne-1,8-diamine (PyED), rapidly cyclizes upon reaction with MgCl2 (t1/2=2.2 h) under ambient conditions.
The key features of this result are the identification of a ligand framework that results in facile Bergman cyclization upon metal-binding activation, and the thermal accessibility of that temperature. We have observed analogous results with Fe(II) and Cu(II), as well as Pt(II) using the same general scaffold (Scheme 1). Of these three metals with tetradentate coordination, the Pt2+ species (R = H) cyclizes almost too quickly, and we have trouble keeping it stable prior to administration. The Cu(II) and the Fe(II) complexes, on the other hand, are more stable and provide a sufficient handling period prior to reaction. Of these, the Fe2+ framework may be more functionally viable because the structure is consistently octahedral even if oxidized, unlike Cu(II) which has wide structural variance across both Cu(I) (tetahedral) and Cu(II)(tetragonal) oxidation states. This makes the Cu-complex utility environment dependent, as reduction will turn off Bergman reactivity completely.
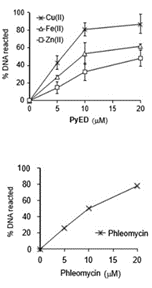 Figure 2. (A)φX174 RFI supercoiled DNA was
Figure 2. (A)φX174 RFI supercoiled DNA was
incubated with PyED or PyBD in the absence
or presence of M(II) for 1 h at 42°C.
Biological Reactivity
While the Cu(II) species has shown the ability to degrade Ab plaques, both Cu(II) and Fe(II) frameworks are functional to degrade DNA at or below the dose rate of Fe-phleomycin within 1 h (Figure 2). The Cu(II) species also possess sufficient rate of reaction to compete with DNA replication and demonstrates G2/M arrest compared with untreated samples, indicating significant DNA damage, while both complexes kill HeLa cells at moderate concentrations. Despite slightly slower reaction kinetics, the Fe2+ complex may be more effective for tumor treatment as it is structurally more predictable and possesses dual threat capability by performing complementary Fenton reactivity

